
Se trata de una enfermedad autoinmune, en la que se ven afectadas las plaquetas, que son la primera línea de defensa contra las hemorragias.
Las plaquetas son células pequeñas y pegajosas, se producen a partir de fragmentos de células grandes llamadas megacariocitos que se originan en la médula ósea (tejido que se encuentra en los huesos largos del cuerpo), salen a la circulación periférica donde ejercen su función principal que es el inicio de la formación del coágulo en los sitios de sangrado. Viven en el cuerpo entre 10-12 días y su valor normal es entre 150000 y 450000 plaquetas por microlitro de sangre.
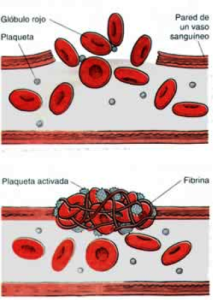 Al sufrir una lesión que afecte un vaso sanguíneo se pierde la continuidad de su pared, dejando salir los glóbulos rojos y el plasma, esto genera un estimulo para que las plaquetas se activen y actúen sobre la lesión formando el coágulo primario.
Al sufrir una lesión que afecte un vaso sanguíneo se pierde la continuidad de su pared, dejando salir los glóbulos rojos y el plasma, esto genera un estimulo para que las plaquetas se activen y actúen sobre la lesión formando el coágulo primario.
Las enfermedades autoinmunes son causadas porque el sistema de defensa del organismo ataca las células propias y las destruye. Aquí, el sistema inmunitario se convierte en el agresor y ataca a partes del cuerpo en vez de protegerlo.
En el caso de la PTI, el organismo detecta a las plaquetas como cuerpos extraños, y las elimina, destrucción que se lleva a cavo en el bazo principalmente y en menor proporción en el hígado.
Con el inicio de la enfermedad aparecen manifestaciones como hematomas espontáneos (sin traumas previos), los cuales suelen ser la manifestación clínica más común, aunque pueden presentarse pequeñas manchas de color púrpura en la piel (petequias) sangrado de cualquier tipo (encías, nasal, tracto gastrointestinal, tracto genitourinario, sistema nervioso central) los cuales dependerán del número de plaquetas. Generalmente el recuento normal disminuye de manera progresiva pero valores mayores de 30000 son suficientes para evitar hemorragias, los valores menores de 10000, se consideran de alto riesgo para sangrados en el sistema nervioso central (intracerebrales) que pueden ser muy graves.
El origen de la PTI no es bien conocido, puesto que se presenta en algunos pacientes previamente sanos y en otros de iguales características no.
Afecta a niños entre dos y diez años, con un pico entre los dos y cinco años, predomina en niñas en todas las edades con una frecuencia de 2-3 niñas por cada niño.
Se ha visto en pacientes quienes presentan unos días o semanas antes del inicio del cuadro, infecciones virales, bacterianas o ha estado expuestos a alguna toxina; se cree que por algunas similitudes que comparten las plaquetas y estos agentes, el sistema de defensa del cuerpo se confunde generando anticuerpos que destruyen el cuerpo extraño y también a las plaquetas.
Por otro lado, también se ha evidenciado la aparición de la enfermedad posterior a aplicación de algunas vacunas entre ellas la mas mencionada es la MMR, pero no se conoce la razón ya que existen niños expuestos a las mismas vacunas que no la desarrollan.
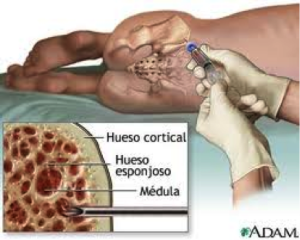
El diagnóstico de La PTI se realiza después de descartar otras causas de trombocitopenia (conteo bajo de plaquetas). No existe un examen específico que confirme el diagnóstico. En ocasiones, cuando los pacientes no responden o no mejoran con el tratamiento instaurado o presentan más de tres episodios de trombocitopenia, se debe descartar alteraciones en la producción de las células precursoras de las plaquetas ( megacariocitos), con un aspirado de medula ósea, que consiste en tomar una muestra de la sangre de un hueso grande y esponjoso como el de la cadera, donde se puede analizar y observar la presencia de los megacariocitos, para verificar que haya buena producción de las plaquetas y descartar alteración de la medula ósea.
La mayoría de los pacientes se manejan ambulatoriamente con observación y signos de alarma, se hace control semanal o bisemanal de plaquetas hasta que estén en valores seguros (mayores de 30000) y luego más espaciado hasta que sea mayor de 150000 plaquetas.
Si se requiere manejo hospitalario en los casos en que el conteo de plaquetas es menor de 10000 es en general menor de 3 días.
El tratamiento de la PTI aguda, depende del número de plaquetas y de las condiciones clínicas del paciente. Generalmente la enfermedad es autolimitada, esto significa que se resuelve sola en un lapso de pocos días, al primer mes el 50% de los pacientes han mejorado y al tercero el 70% de los casos han recuperado el nivel normal de plaquetas; el 30% restante se resolverá hasta el sexto mes y los que no están en este grupo, pasaran a padecer una PTI crónica.
En cuanto al número de plaquetas, los pacientes con un recuento mayor a 30000 no requieren tratamiento ya que el riesgo de sangrado es menor del 3%; si el recuento es menor 10000 se inicia tratamiento.
En el caso de que el paciente presente algún tipo de sangrado moderado o severo se inicia tratamiento independiente del número de plaquetas.
Esteroides: Aumentan la producción de plaquetas, disminuye la producción anticuerpos por tanto disminuye la destrucción plaquetaria. Se administra en tres regímenes diferentes: 1. Prednisona vía oral por 14 días + 1 semana de desmonte (disminución progresiva de la dosis). 2. Prednisona vía oral por 4 días. 3. Metilprednisolona intravenosa por 3 días. El uso de uno u otro régimen depende del juicio clínico del médico tratante. Todos los medicamentos tienen efectos 2rios en el caso de los esteroides se pueden presentar cambios en la conducta, alteraciones del sueño, aumento de apetito y por consiguiente de peso, alteraciones del comportamiento especialmente con ciclos repetidos y largos de esteroides.
Otras alternativas son el uso de Inmunoglobulina humana o Inmunoglobulina anti D, estos son anticuerpos similares a los producidos por el cuerpo que bloquean las defensas dirigidas contra las plaquetas evitando su destrucción. Como efectos secundarios puede presentarse fiebre, cefalea, anemia hemolítica. Como se mencionó anteriormente el uso de uno u otro medicamento depende de las condiciones de cada paciente y del criterio del médico tratante.
La transfusión de plaquetas solo está indicada en presencia de sangrado intracerebral ó una hemorragia severa que amenace la vida.
 Otra opción para pacientes que no responden a las terapias descritas o cursan con una PTI crónica sintomática es la esplenectomía (extirpación quirúrgica del Bazo órgano en el cual se lleva a cavo la destrucción de la mayor parte de las plaquetas durante el curso de la enfermedad), sin embargo esta decisión es netamente medica.
Otra opción para pacientes que no responden a las terapias descritas o cursan con una PTI crónica sintomática es la esplenectomía (extirpación quirúrgica del Bazo órgano en el cual se lleva a cavo la destrucción de la mayor parte de las plaquetas durante el curso de la enfermedad), sin embargo esta decisión es netamente medica.
Cuando existen presentaciones atípicas de la enfermedad o síntomas predominantes como cansancio, hipoactividad, fiebre prolongada, dolor óseo o articular, anemia, disminución en el recuento de neutrofilos (células de defensa) y PTI crónica (trombocotopenia con recuento de plaquetas menor de 150000 después de 6 meses del diagnostico) está indicado realizar aspirado de medula ósea para descartar otros diagnósticos como Leucemia.
Agradecimientos a la Dra.:
Diana Isabel Bravo Cerón, Residente de Pediatría, Universidad del CES- FVL por construir esta información.
Bibliografía
– Steuber CP. Clinical manifestations and diagnosis of immune (idiopathic) thrombocytopenic purpura in children. Uptodate. 2008.
– Steuber CP. Treatment and prognosis of immune (idiopathic) thrombocytopenic purpura in children. Uptodate. 2008.
– Pediatr Clin N Am 55 (2008) 393–420.
– Pediatrics in Review Vol. 21 No. 3 March 2000.